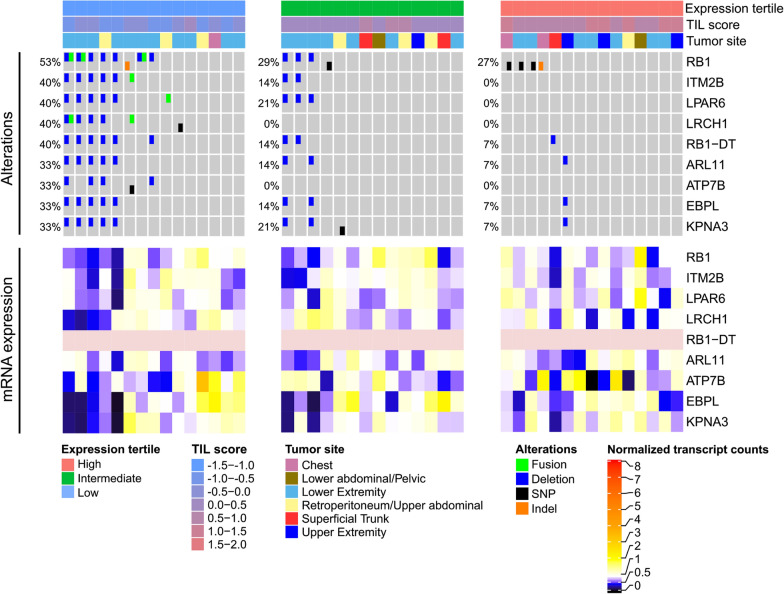
Impact of genetic aberrations on the Tumor Microenvironment (TME)
We aim to investigate how genetic aberrations in cancer cells influence the composition and functional state of the Tumor Microenvironment (TME). Such aberrations may affect the recruitment, abundance, or activation state of various cell types within the TME, ultimately shaping the tumor’s immunogenicity. For instance, certain genetic changes can promote infiltration by Tumor-Infiltrating Lymphocytes (TILs), resulting in so-called “hot” tumors, which are recognized and targeted by the immune system. In contrast, “cold” tumors lack significant immune cell infiltration and remain largely undetected by the host’s immune response. These differences are clinically relevant, as patients with hot tumors generally experience better outcomes and may respond more favourably to immunotherapies.
Relevant publications:
Deconvolution of sarcoma methylomes reveals varying degrees of immune cell infiltrates with association to genomic aberrations
J Transl Med. 2022 Apr 2;20(1):152. doi: 10.1186/s12967-021-02858-7
Chronic Chromosome Instability Induced by Plk1 Results in Immune Suppression in Breast Cancer
Cell Reports. 2023 Dec 26;42(12):113266. doi: 10.1186/s12967-021-02858-7
Tissue Tregs function in health and cancer
We investigate how regulatory T cells (Tregs) adapt to specific tissues. Tissue Tregs not only suppress other immune cells, but they can also acquire tissue-adapted states linked to immune homeostasis and tissue repair, and these states overlap with features seen in tumor-associated Tregs. We focus on the molecular imprinting of tissue Tregs: we study epigenetic and chromatin programs that distinguish Tregs within different tissues and blood.
Relevant publications:
Polyamines regulate adaptive antitumor immunity by functional specialization of regulatory T cells
Immunity . 2025 Aug 12;58(8):2019-2034.e11. doi: 10.1016/j.immuni.2025.07.007.
DNA hypomethylation traits define human regulatory T cells in cutaneous tissue and identify their blood recirculating counterparts.
Nat Immunol . 2025 Aug;26(8):1315-1328. doi: 10.1038/s41590-025-02210-x.
Single-cell chromatin accessibility and transposable element landscapes reveal shared features of tissue-residing immune cells
Immunity . 2024 Aug 13;57(8):1975-1993.e10. doi: 10.1016/j.immuni.2024.06.015.
Single-cell chromatin accessibility landscape identifies tissue repair program in human regulatory T cells
Immunity . 2021 Apr 13;54(4):702-720.e17. doi: 10.1016/j.immuni.2021.03.007.
Precursors for Nonlymphoid-Tissue Treg Cells Reside in Secondary Lymphoid Organs and Are Programmed by the Transcription Factor BATF
Immunity . 2020 Feb 18;52(2):295-312.e11. doi: 10.1016/j.immuni.2019.12.002.
Genome-wide DNA-methylation landscape defines specialization of regulatory T cells in tissues
Nat Immunol . 2017 Oct;18(10):1160-1172. doi: 10.1038/ni.3799.
tissueTreg BioConductor (Data Package)

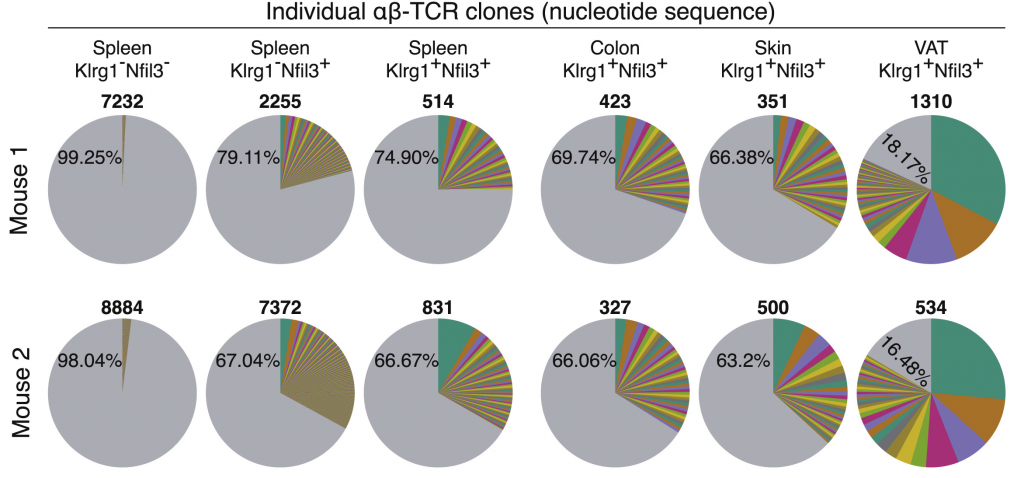
Leveraging TCRs to Understand T Cell Clonality Dynamics in Health and Disease
T Cell Receptors (TCRs) provide a unique molecular barcode, enabling us to track individual T cell clones across time, tissues, and disease states. By analyzing TCR repertoires, we uncover how T cells expand, persist, and adapt—whether in response to infection, within the tumor microenvironment, or following vaccination. Our research aims to decode these clonality dynamics to better understand immune activation and to map T cell migration across tissues and circulation.
Relevant publications:
Precursors for Nonlymphoid-Tissue Treg Cells Reside in Secondary Lymphoid Organs and Are Programmed by the Transcription Factor BATF
Immunity. 2020 Feb 18;52(2):295-312.e11. doi: 10.1016/j.immuni.2019.12.002
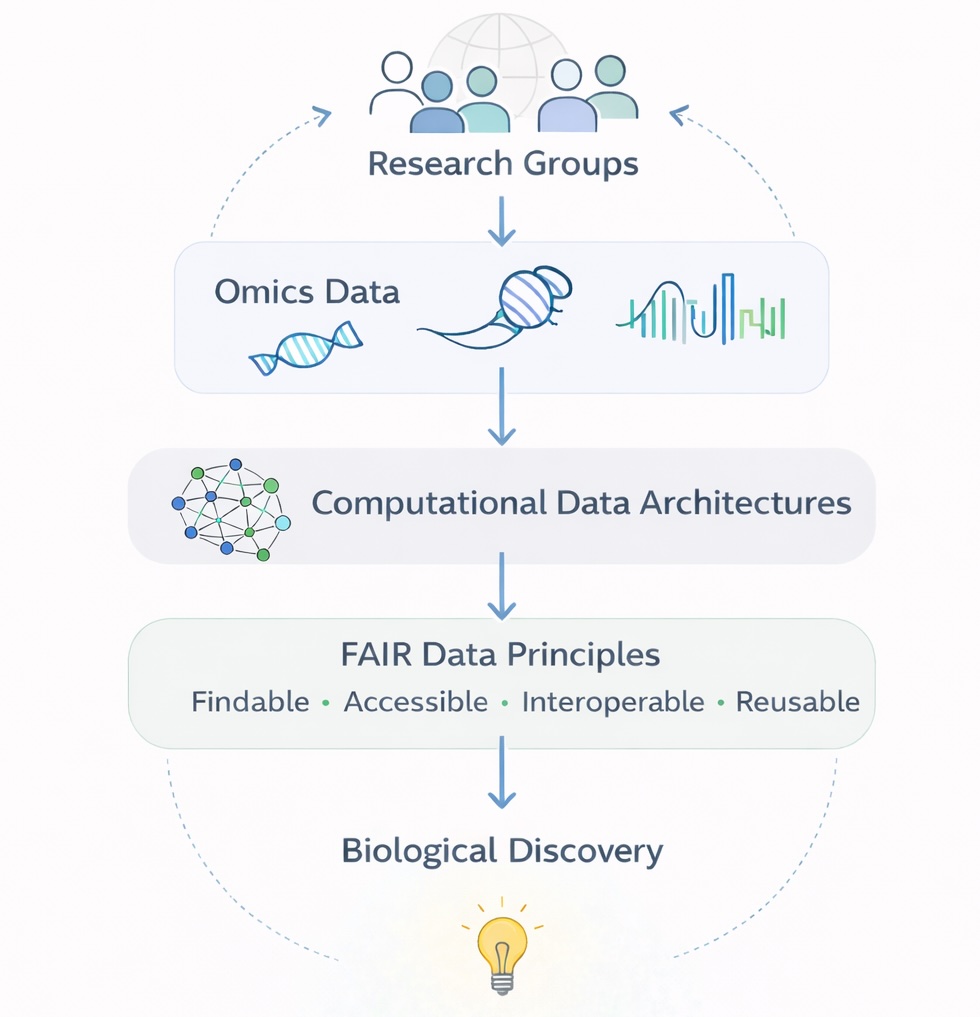
Computational Data Architectures for Omics Research
Modern immunology increasingly relies on large-scale sequencing and multi-omics technologies that generate complex, high-dimensional datasets. Extracting biological insight from these data requires not only advanced analytical approaches but also robust and reproducible computational infrastructures.
Our lab develops computational frameworks that enable scalable analysis of large omics datasets. This includes automated pipelines for sequencing data processing, integration of multiple data modalities, and computational environments designed to ensure data integrity, reproducibility, and compliance with FAIR data principles (Findable, Accessible, Interoperable, Reusable). We also develop solutions that facilitate secure data exchange and collaboration between research groups.
Our work builds on experience from large international genomics initiatives. Members of the lab previously contributed to projects of the International Cancer Genome Consortium (ICGC), including German ICGC studies on early-onset prostate cancer (EOPC), multiple myeloma (MMML), and pediatric glioblastomas (GBMs). The lab has also participated in the DEEP (German Epigenome Programme) consortium generating reference epigenome maps.
Relevant publications:
Epigenomic Profiling of Human CD4+ T Cells Supports a Linear Differentiation Model and Highlights Molecular Regulators of Memory Development
Immunity. 2016 Nov 15;45(5):1148-1161. doi: 10.1016/j.immuni.2016.10.022
The whole-genome landscape of medulloblastoma subtypes
Nature. 2017 Jul 19;547(7663):311-317. doi: 10.1038/nature22973
Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma
Nat Genet. 2013 Aug;45(8):927-32. doi: 10.1038/ng.2682